Project description
Link to myc Chip-seq data: myc
Link to control Chip-seq data: control
Raw data downloding and reads mapping
Raw data were downloaded by the following commands:
wget https://www.encodeproject.org/files/ENCFF001NQP/@@download/ENCFF001NQP.fastq.gz
wget https://www.encodeproject.org/files/ENCFF001NQQ/@@download/ENCFF001NQQ.fastq.gz
wget https://www.encodeproject.org/files/ENCFF001NCH/@@download/ENCFF001NCH.fastq.gz
wget https://www.encodeproject.org/files/ENCFF001NCF/@@download/ENCFF001NCF.fastq.gz
mv ENCFF001NQP.fastq.gz rep1.fastq.gz
mv ENCFF001NQQ.fastq.gz rep2.fastq.gz
mv ENCFF001NCH.fastq.gz input1.fastq.gz
mv ENCFF001NCF.fastq.gz input2.fastq.gz
fqSample <- FastqSampler("/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/raw_data/rep1.fastq.gz", n = 10^6)
fastq <- yield(fqSample)
fastq
class: ShortReadQ
length: 1000000 reads; width: 36 cycles
Raw ChIPseq data QC
We can access different metrics of fastq files by following functions:sread() - Retrieve sequence of reads.
quality() - Retrieve quality of reads as ASCII scores.
id() - Retrieve IDs of reads.
readSequences <- sread(fastq)
readQuality <- quality(fastq)
readIDs <- id(fastq)
readSequences
DNAStringSet object of length 1000000:
width seq
[1] 36 ACAGTAGTCTTCCGGTCAAGAGCTACACGGGTTGTG
[2] 36 TGTGTGTATGTCTGCCTGCCAGCAGAGGGAACCAGA
[3] 36 CTGATTCAAACTCTGTTCTGTCAGCTTCTGACTGAA
[4] 36 AGATACCATATAATGAACCCACGAAAACAATCCGGT
[5] 36 CTAAAATAACCAAAGGACTTGTCACATCTATGTAAA
... ... ...
[999996] 36 GTTTATAAACAGCATTGTACGTTACCCAGGACCTGC
[999997] 36 AAACAGGAGGAAGGGAGGATTGAGGGACCGCACAGA
[999998] 36 CAACAGGTTTGATCTTCACGCTCGGCTTATTCTTGC
[999999] 36 GTGACTTTACCCGCTTTATCTCTGGACTCCAGAGTT
[1000000] 36 TTTCCTACTTTACAGCATAGATGATGAAAAACACAA
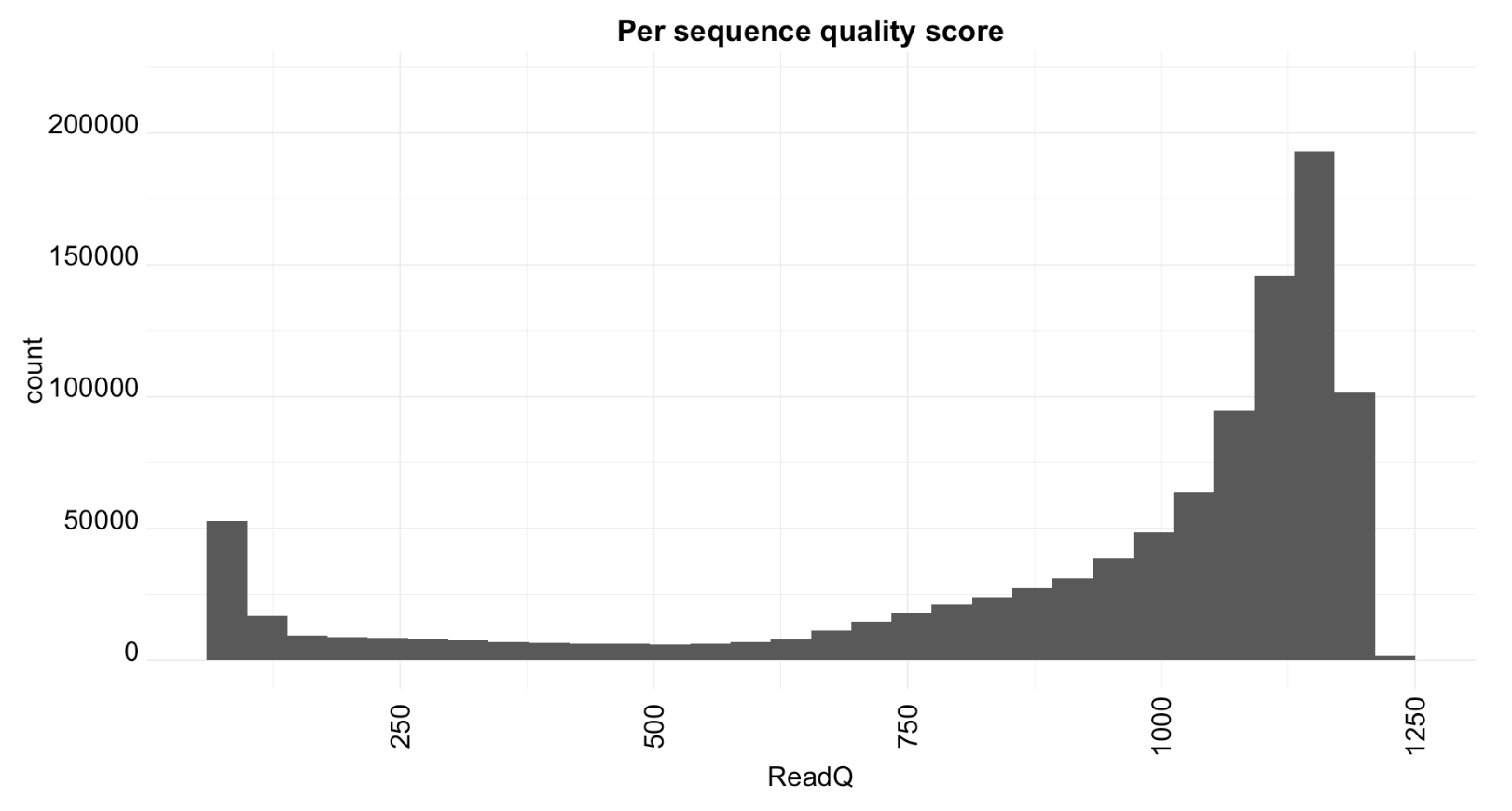
Histogram of quality scores.
library(ggplot2)
library(repr)
options(repr.plot.width=15, repr.plot.height=8)
toPlot <- data.frame(ReadQ = readQualities)
ggplot(toPlot, aes(x = ReadQ)) + geom_histogram(bins=30) + theme_minimal()+
theme(axis.text.x = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 20, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
plot.title = element_text(size=22, hjust = .5, vjust = .5, face="bold"))+
ylim(0, 220000)+ggtitle("Per sequence quality score")
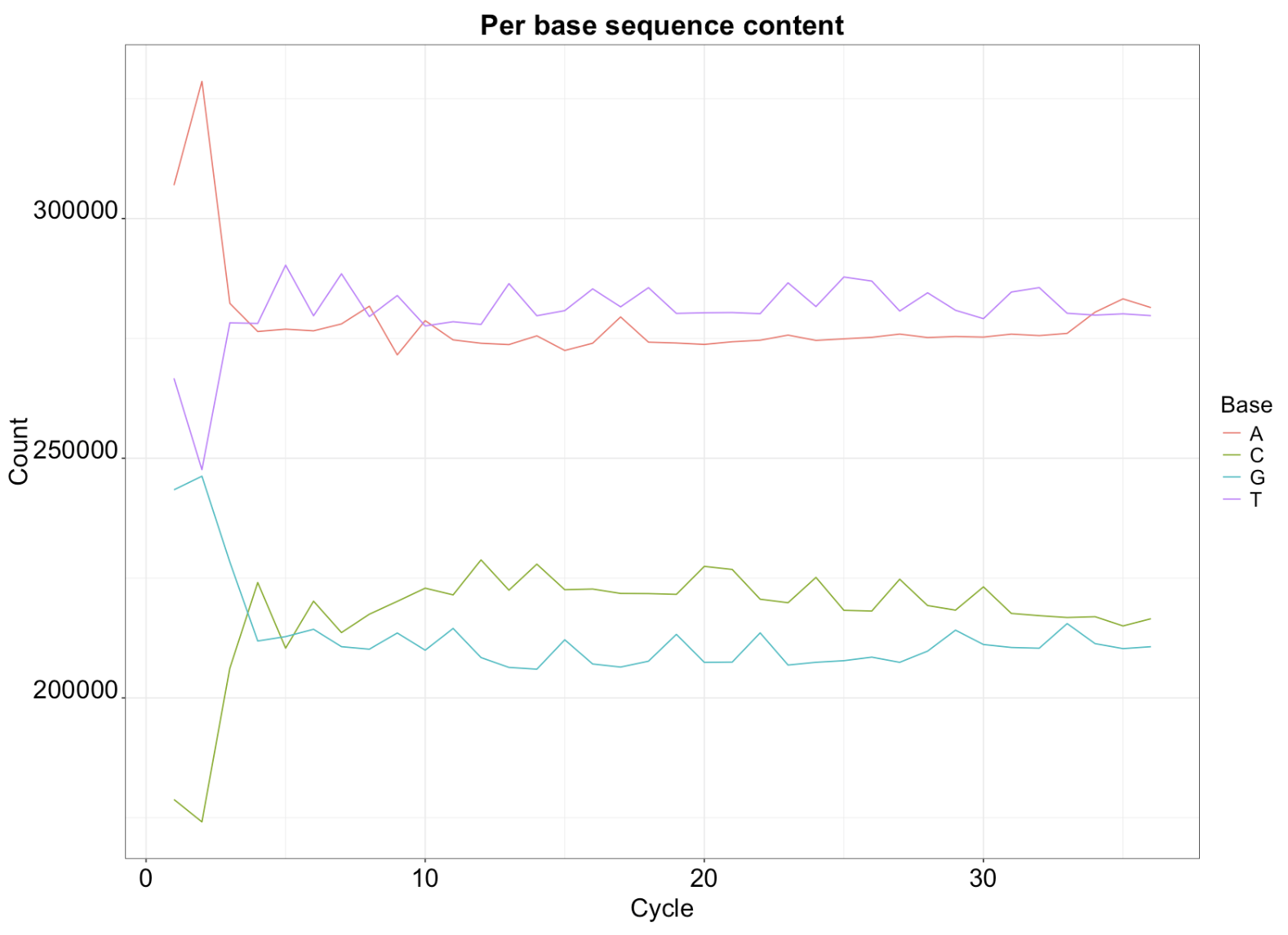
Visualizing base occurrence over positions in reads
readSequences_AlpbyCycle <- alphabetByCycle(readSequences)
readSequences_AlpbyCycle[1:4, 1:10]
A matrix: 4 × 10 of type int
A 306954 328614 282305 276432 276920 276570 278020 281716 271557 278703
C 178805 174129 206175 224071 210374 220172 213623 217461 220140 222889
G 243413 246240 228349 211866 212787 214306 210685 210163 213540 209952
T 266665 247611 278246 278121 290270 279718 288472 279555 283949 277603
AFreq <- readSequences_AlpbyCycle["A", ]
CFreq <- readSequences_AlpbyCycle["C", ]
GFreq <- readSequences_AlpbyCycle["G", ]
TFreq <- readSequences_AlpbyCycle["T", ]
toPlot <- data.frame(Count = c(AFreq, CFreq, GFreq, TFreq), Cycle = rep(1:36, 4),
Base = rep(c("A", "C", "G", "T"), each = 36))
ggplot(toPlot, aes(y = Count, x = Cycle, colour = Base)) + geom_line() + theme_bw()+
theme(axis.text.x = element_text(color = "black", size = 20, angle =0, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 20, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
plot.title = element_text(size=22, hjust = .5, vjust = .5, face="bold"))+
theme(
legend.text = element_text(size = 16),
legend.title = element_text(size = 18))+ggtitle("Per base sequence content")
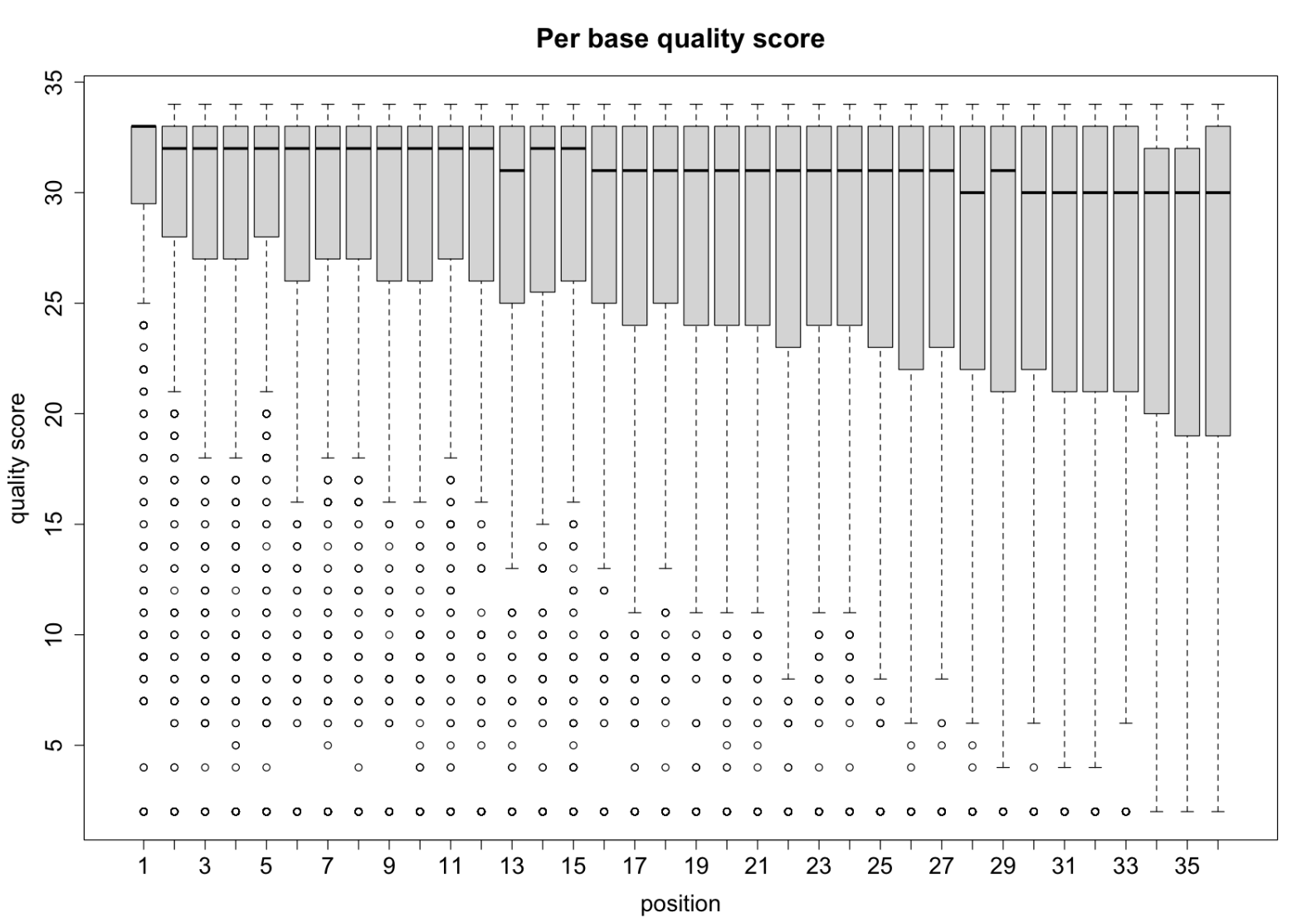
Per base quality score
options(repr.plot.width=14, repr.plot.height=10)
graphics::boxplot(qualAsMatrix[1:1000, ], xlab="position", ylab="quality score", main="Per base quality score", cex.axis=1.5, cex.lab=1.5, cex.main=1.75)
Filtering out the low quality reads
Low quality reads will be filtered out based on following criteria:Quality score more than 300
The number of N in the read is less than 10
First, path to filterd reads was created:
files <- list.files(path="/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data",pattern="*.fastq.gz")
files_filt<- paste("filt", files, sep = "_")
path_filt<-paste0("/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/", files_filt)
path_filt
'/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/filt_input1.fastq.gz'
'/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/filt_input2.fastq.gz'
'/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/filt_rep1.fastq.gz'
'/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/filt_rep2.fastq.gz'
mylist <- lapply(paste0("/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/",files), function(x) FastqStreamer(x, n = 1e+05))
mylist
[[1]]
class: FastqStreamer
file: input1.fastq.gz
status: n=100000 current=0 added=0 total=0 buffer=0
[[2]]
class: FastqStreamer
file: input2.fastq.gz
status: n=100000 current=0 added=0 total=0 buffer=0
[[3]]
class: FastqStreamer
file: rep1.fastq.gz
status: n=100000 current=0 added=0 total=0 buffer=0
[[4]]
class: FastqStreamer
file: rep2.fastq.gz
status: n=100000 current=0 added=0 total=0 buffer=0
k=1
for (i in mylist){
TotalReads <- 0
TotalReadsFilt <- 0
while (length(fq <- yield(i)) > 0) {
TotalReads <- TotalReads + length(fq)
filt1 <- fq[alphabetScore(fq) > 300]
filt2 <- filt1[alphabetFrequency(sread(filt1))[, "N"] < 10]
TotalReadsFilt <- TotalReadsFilt + length(filt2)
writeFastq(filt2, path_filt[k], mode = "a")
k=k+1
}
}
Reads mapping and peaks calling
Reads mapping and peaks calling were performed in zsh shell.bowtie2 was used for reads mapping
macs2 was used for peak calling
index=/Users/alexeyefanov/Desktop/Programming/Bioconductor/refgenomes/mouse_index
filt=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/filtered
reference=/Users/alexeyefanov/Desktop/Programming/Bioconductor/refgenomes/BSgenome.Mmusculus.UCSC.mm10.mainChrs.fa
sam=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam
bowtie2-build -f $reference $index/index
gunzip -k $filt/*.gz
cd $sam
for sample in $filt/*.fastq; do
base=$(basename $sample .fastq)
bowtie2 -x $index/index -fr -q $sample -p 12 -S $base.sam
done
for sample in $sam/*.bam;do
base=$(basename $sample .bam)
samtools view -S -b -o $base.bam $base.sam
samtools sort -o ${base}_sorted.bam ${base}.bam
samtools index ${base}_sorted.bam
done
input1=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_input1_sorted.bam
input2=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_input2_sorted.bam
rep1=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_rep1_sorted.bam
rep2=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_rep2_sorted.bam
peak=/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/peaks
#conda activate MACS
macs2 callpeak -t $rep1 $rep2 -c $input1 $input2 --name mel --outdir $peak
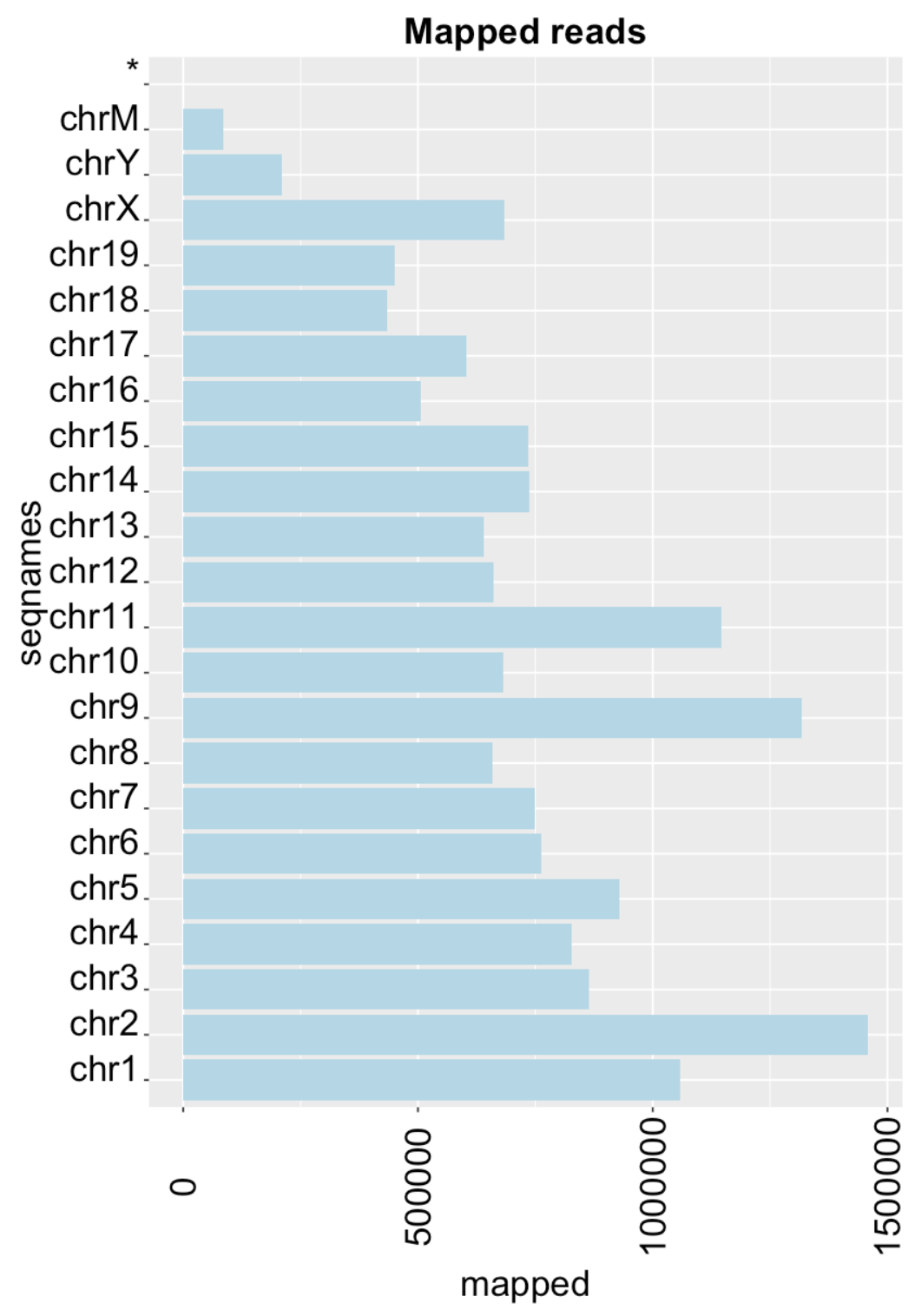
Mapped reads
library(repr)
options(repr.plot.width=9, repr.plot.height=15)
mappedReads <- idxstatsBam("/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_rep1_sorted.bam")
TotalMapped <- sum(mappedReads[, "mapped"])
ggplot(mappedReads, aes(x = seqnames, y = mapped)) + geom_bar(stat = "identity") +
coord_flip()+
theme(axis.text.x = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 15, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 15, angle = 90, hjust = .5, vjust = .5, face = "plain"))
Creating BigWig and visualizing mapped reads usin IGV
The bigWig format is used to display genomics data in such tools as the UCSC Genome Browser, and The Integrative Genomics Viewer (IGV) as a graph.
BigWig <- coverage("/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_rep1_sorted.bam", weight = (10^6)/TotalMapped)
library(rtracklayer)
export.bw(BigWig, con = "/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/sam/filt_rep1_weighted1.bw")
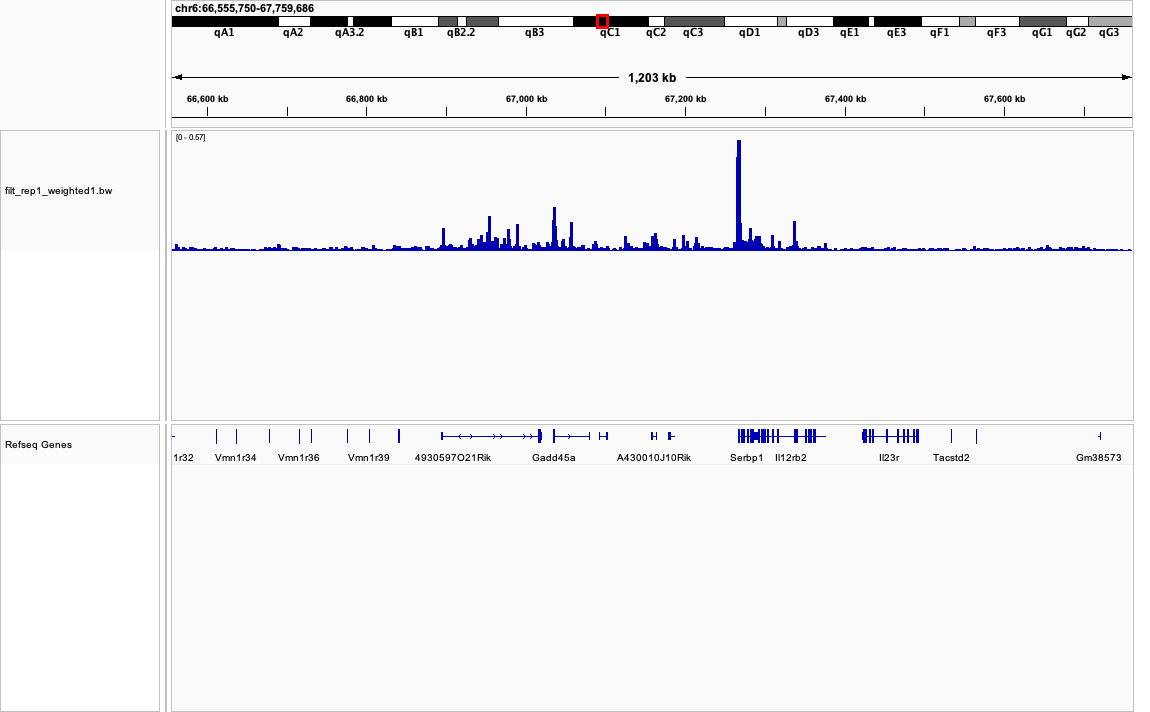
Downloading output of macs2
macsPeaks <- "/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/peaks/mel_peaks.xls"
macsPeaks_DF <- read.delim(macsPeaks, comment.char = "#")
macsPeaks_DF[1:2, ]
A data.frame: 2 × 10
chr start end length abs_summit pileup X.log10.pvalue. fold_enrichment X.log10.qvalue. name
1 chr1 4785344 4785933 590 4785600 29.56 14.49350 5.46755 11.17909 mel_peak_1
2 chr1 5083013 5083207 195 5083083 35.06 11.00655 3.60638 7.87681 mel_peak_2
Converting MACS peaks to GRanges object
library(GenomicRanges)
macsPeaks_GR <- GRanges(seqnames = macsPeaks_DF[, "chr"], IRanges(macsPeaks_DF[,
"start"], macsPeaks_DF[, "end"]))
macsPeaks_GR
GRanges object with 26241 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr1 4785344-4785933 *
[2] chr1 5083013-5083207 *
[3] chr1 5677542-5677817 *
[4] chr1 6012559-6012715 *
[5] chr1 7397544-7398143 *
... ... ... ...
[26237] chrY 58550402-58550723 *
[26238] chrY 60505268-60505555 *
[26239] chrY 89932367-89932501 *
[26240] chrY 90818621-90818899 *
[26241] chrY 90824605-90824835 *
-------
seqinfo: 21 sequences from an unspecified genome; no seqlengths
table(seqnames(macsPeaks_GR))
chr1 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr2 chr3
1506 1332 2635 1018 1007 790 1167 852 1304 700 961 1924 1357
chr4 chr5 chr6 chr7 chr8 chr9 chrX chrY
1567 1840 1261 1602 1197 1489 716 12
Adding metadata
mcols(macsPeaks_GR) <- macsPeaks_DF[, c("abs_summit", "fold_enrichment")]
macsPeaks_GR
Filtering out regions overlaping with blacklisted regions
Blacklisted regions are regions showing anomalous read mapping to them.
toBlkList <- "/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/blacklist/ENCFF547METmm10.bed"
blkList <- import.bed(toBlkList)
macsPeaks_GR <- macsPeaks_GR[!macsPeaks_GR %over% blkList]
macsPeaks_GR
GRanges object with 26237 ranges and 2 metadata columns:
seqnames ranges strand | abs_summit fold_enrichment
|
[1] chr1 4785344-4785933 * | 4785600 5.46755
[2] chr1 5083013-5083207 * | 5083083 3.60638
[3] chr1 5677542-5677817 * | 5677571 5.65644
[4] chr1 6012559-6012715 * | 6012601 5.69169
[5] chr1 7397544-7398143 * | 7397789 8.75376
... ... ... ... . ... ...
[26233] chrY 58550402-58550723 * | 58550543 17.33837
[26234] chrY 60505268-60505555 * | 60505432 8.35447
[26235] chrY 89932367-89932501 * | 89932468 5.10398
[26236] chrY 90818621-90818899 * | 90818649 3.21887
[26237] chrY 90824605-90824835 * | 90824624 2.55565
-------
seqinfo: 21 sequences from an unspecified genome; no seqlengths
Peak annotation
The peakAnno object contains the information on annotation of individual peaks to genes.
peakAnno <- annotatePeak(macsPeaks_GR, tssRegion = c(-500, 500), TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene,
annoDb = "org.Mm.eg.db")
peakAnno
Annotated peaks generated by ChIPseeker
26237/26237 peaks were annotated
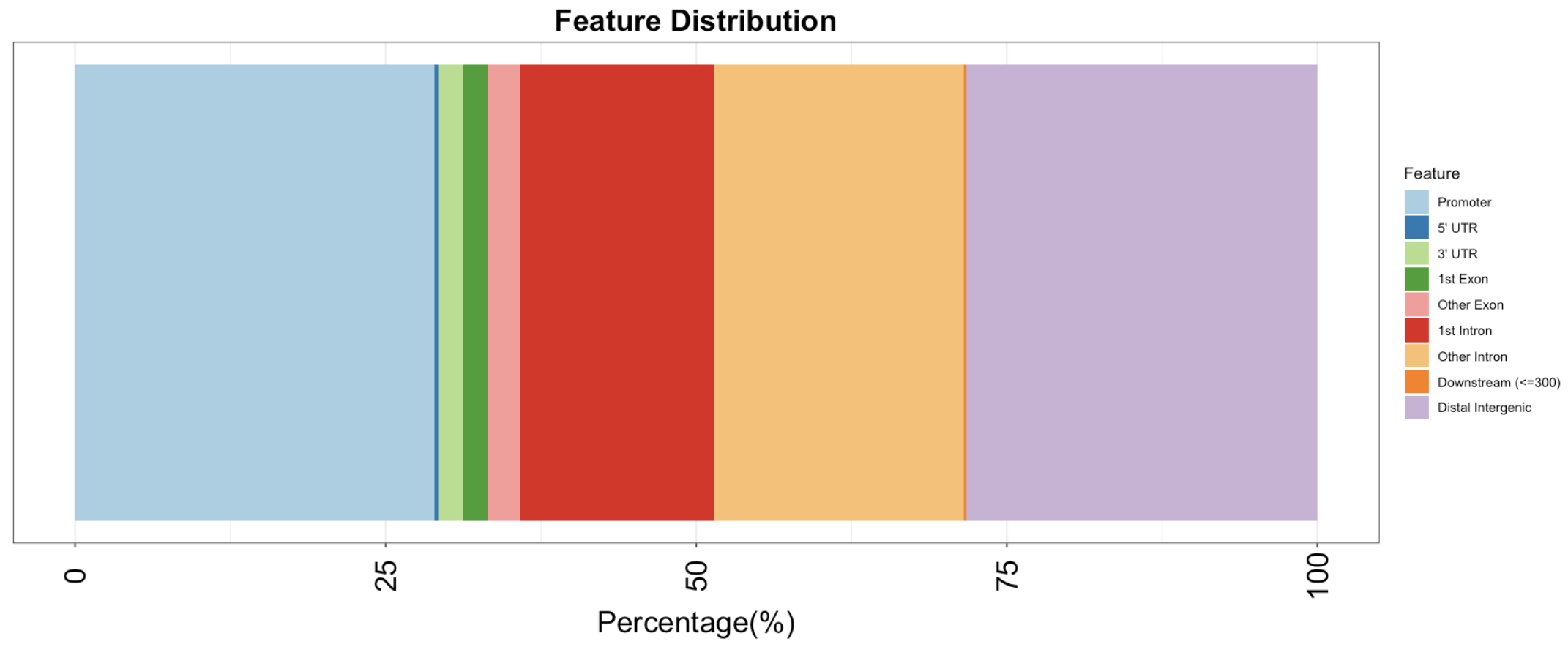
Genomic Annotation Summary:
Feature Frequency
9 Promoter 28.9514807
4 5' UTR 0.3163471
3 3' UTR 1.9781225
1 1st Exon 2.0048024
7 Other Exon 2.5803255
2 1st Intron 15.5772383
8 Other Intron 20.1433091
6 Downstream (<=300) 0.2210619
5 Distal Intergenic 28.2273126
Converting the peakAnno object to GRanges and Data frame objects
peakAnno_GR <- as.GRanges(peakAnno)
peakAnno_DF <- as.data.frame(peakAnno)
Visualizing of peak annotation
options(repr.plot.width=15, repr.plot.height=6)
plotAnnoBar(peakAnno)+
theme(axis.text.x = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 20, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
plot.title = element_text(size=20, hjust = .5, vjust = .5, face="bold"))
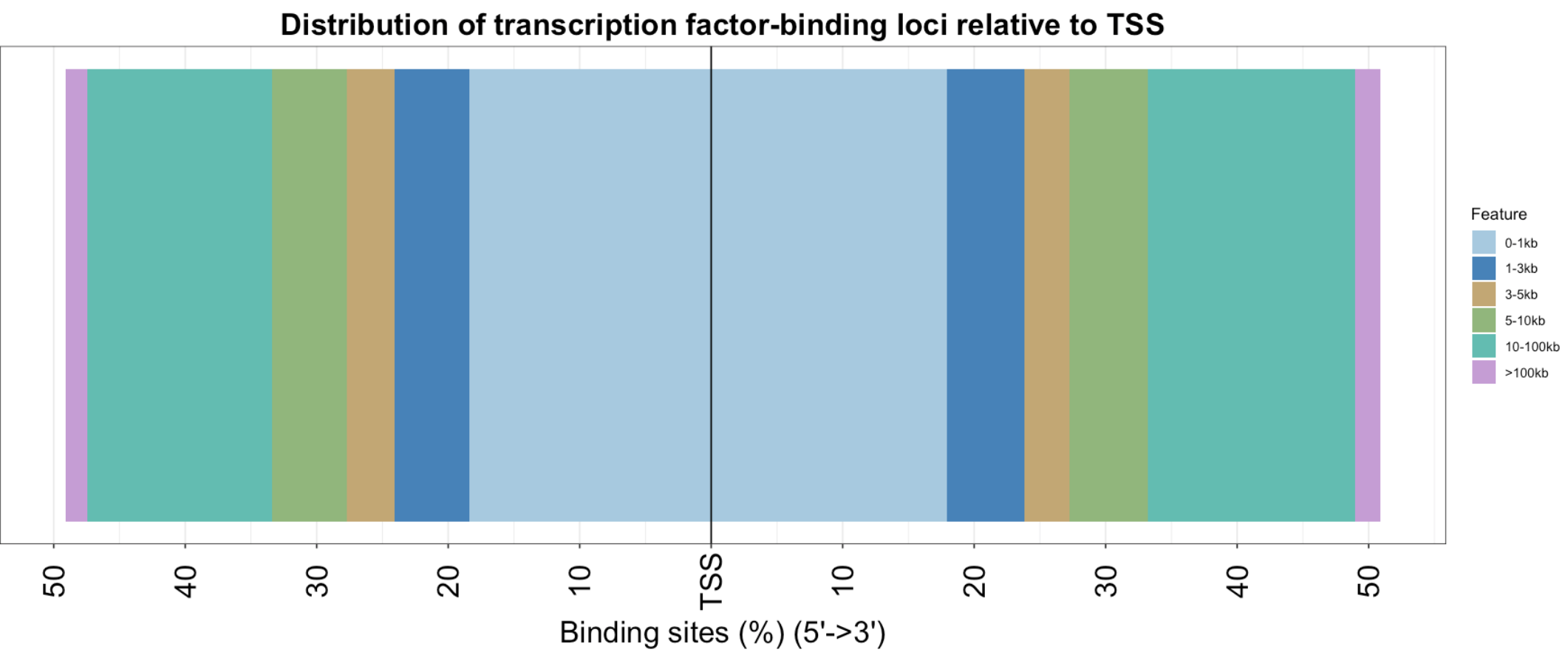
plotDistToTSS(peakAnno)+theme(axis.text.x = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 20, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
plot.title = element_text(size=20, hjust = .5, vjust = .5, face="bold"))
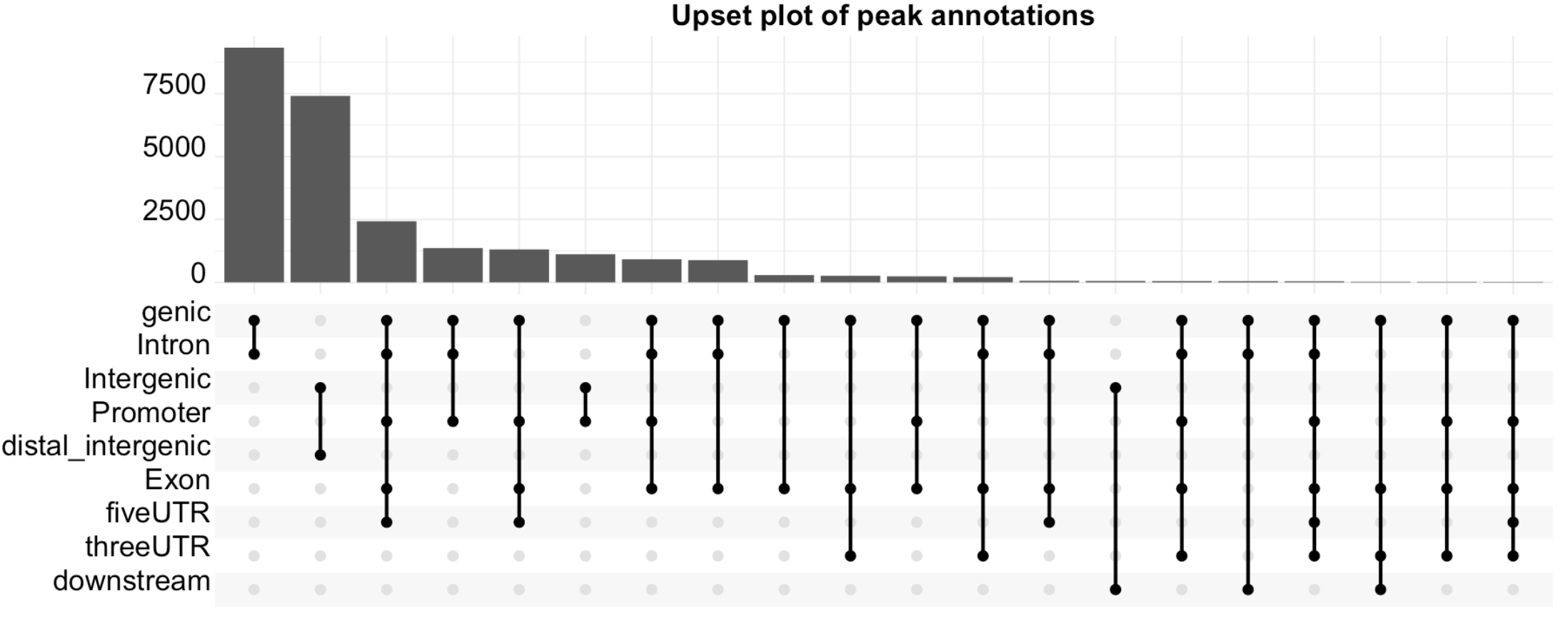
upsetplot(peakAnno, vennpie = F)+theme(axis.text.x = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
axis.text.y = element_text(color = "black", size = 20, angle = 0, hjust = 1, vjust = 0, face = "plain"),
axis.title.x = element_text(color = "black", size = 20, angle = 0, hjust = .5, vjust = 0, face = "plain"),
axis.title.y = element_text(color = "black", size = 20, angle = 90, hjust = .5, vjust = .5, face = "plain"),
plot.title = element_text(size=20, hjust = .5, vjust = .5, face="bold"))+ggtitle("Upset plot of peak annotations")
Annotation of peaks to genes
Since transcription factors, as suggested in name, may regulate the transcription of their target genes, we used the ChIPseeker package to associate our peaks, representing potential transcription factor binding events, to their overlapping or closest mm10 genes.
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
library(ChIPseeker)
peakAnno <- annotatePeak(macsPeaks_GR, tssRegion=c(-1000, 1000),
TxDb=TxDb.Mmusculus.UCSC.mm10.knownGene,
annoDb="org.Mm.eg.db")
Extracting sequences under regions
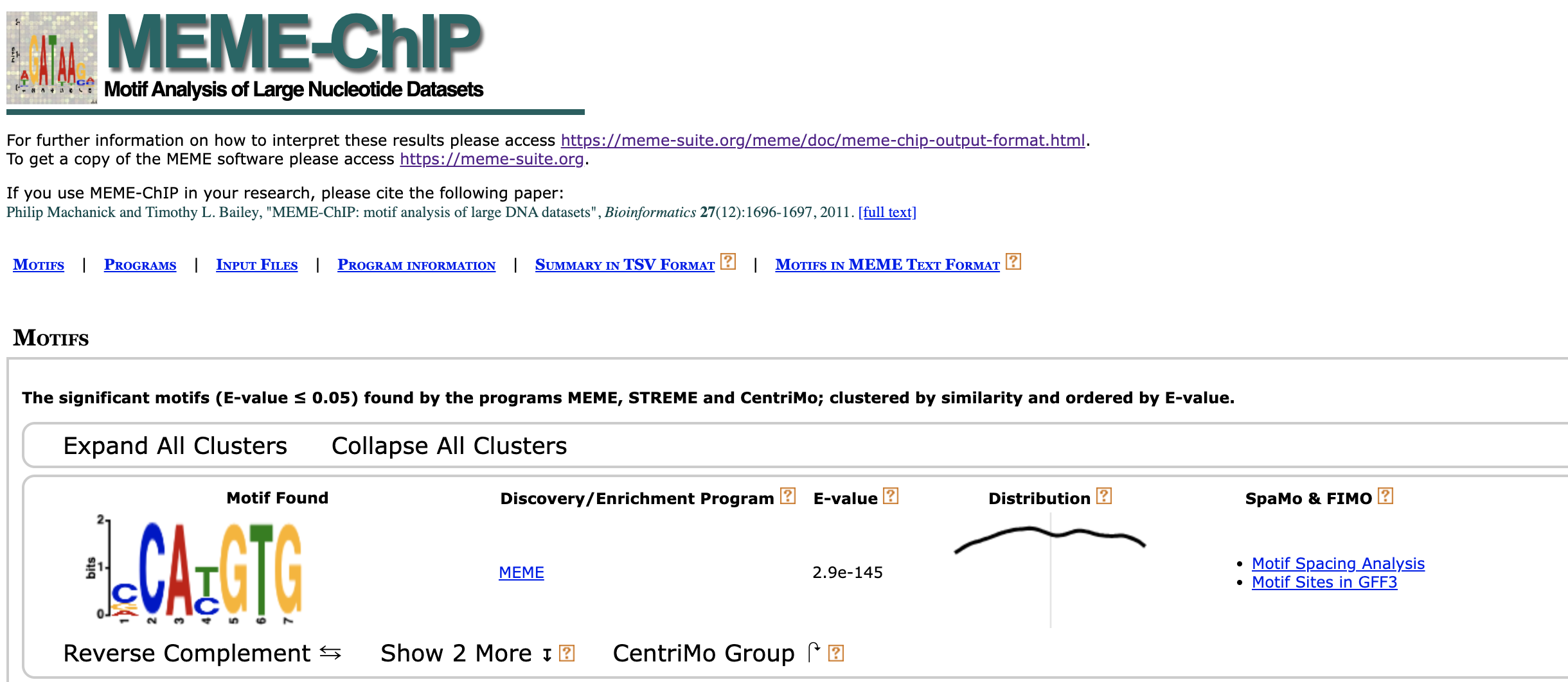
The motif for the ChIP-ed transcription factor should near the summit (the highest point) of a peak. MEME-ChIP will trim our peaks to a common length internally if sequences are of different length. It is best therefore to provide a peak set resized to a common length.
macsSummits_GR <- GRanges(seqnames(macsPeaks_GR), IRanges(macsPeaks_GR$abs_summit,
macsPeaks_GR$abs_summit), score = macsPeaks_GR$fold_enrichment)
macsSummits_GR <- resize(macsSummits_GR, 100, fix = "center")
macsSummits_GR
GRanges object with 26237 ranges and 1 metadata column:
seqnames ranges strand | score
|
[1] chr1 4785550-4785649 * | 5.46755
[2] chr1 5083033-5083132 * | 3.60638
[3] chr1 5677521-5677620 * | 5.65644
[4] chr1 6012551-6012650 * | 5.69169
[5] chr1 7397739-7397838 * | 8.75376
... ... ... ... . ...
[26233] chrY 58550493-58550592 * | 17.33837
[26234] chrY 60505382-60505481 * | 8.35447
[26235] chrY 89932418-89932517 * | 5.10398
[26236] chrY 90818599-90818698 * | 3.21887
[26237] chrY 90824574-90824673 * | 2.55565
-------
seqinfo: 21 sequences from an unspecified genome; no seqlengths
Extracting sequences under regions
peaksSequences <- getSeq(BSgenome.Mmusculus.UCSC.mm10, macsSummits_GR)
names(peaksSequences) <- paste0(seqnames(macsSummits_GR), ":", start(macsSummits_GR),
"-", end(macsSummits_GR))
peaksSequences[1:2, ]
DNAStringSet object of length 2:
width seq names
[1] 100 CCGCACCGCCGCCAAGCGTTTAC...GGACCGCAGCAGGTCCAGGCTGG chr1:4785550-4785649
[2] 100 TTTGGTGCGCGGTCCAGCAGTTT...CAGACGCTGCTGTCACTTCGTAG chr1:5083033-5083132
Saving the peaksSequences object as meme.fa
The meme.fa file will be feeded into MEME-Chip (the comprehensive motif analysis (including motif discovery) tool)
writeXStringSet(peaksSequences, file ="/Users/alexeyefanov/Desktop/Programming/Bioconductor/ChiP-seq/MEL/data/meme.fa").
meme-chip -maxw 10 -minw 5 -o results meme.fa
Back to bioinformatics project page: Back to bioinformatics project page
Back to home: Back to home