Project description
Link to the paper: link
In that paper, authors investigated the role HMGNs in white adipocyte browning. They employed scRNAseq to achieve that goal. WT mice (control) were compared with DKO mice. DKO mice are genetically modified mice that are lacking HMGN1 and HMGN2 proteins. Two bilogical replicates of each line were used at two time points-0 days and 6 days. Totally 8 samples were used in that study. Table below shows sample names and experiment conditions:
| Sample | Species | Condition | Time Point | Replicate | Status |
|---|---|---|---|---|---|
| W10 | Mouse | WT | 0 | 1 | Preadipocyte |
| W20 | Mouse | WT | 0 | 2 | Preadipocyte |
| D10 | Mouse | DKO | 0 | 1 | Preadipocyte |
| D20 | Mouse | DKO | 0 | 2 | Preadipocyte |
| W16 | Mouse | WT | 6 | 1 | Adipocytes |
| W26 | Mouse | WT | 6 | 2 | Adipocytes |
| D16 | Mouse | DKO | 6 | 1 | Adipocytes |
| D26 | Mouse | DKO | 6 | 2 | Adipocyte |
Raw data downloding and reads mapping
scRNAseq sra files (PRJNA796303) can be accessed at link to raw data
The tsv table was downloaded from ENA: PRJNA796303_tsv.txt. Links to sra files were extracted from that file by code:
awk 'NR>1{print $9}' PRJNA796303_tsv.txt | awk '{gsub(/\;/,"\n")};{print $0}' >download3.sh
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/087/SRR17555087
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/088/SRR17555088
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/090/SRR17555090
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/083/SRR17555083
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/084/SRR17555084
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/086/SRR17555086
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/089/SRR17555089
ftp.sra.ebi.ac.uk/vol1/srr/SRR175/085/SRR17555085
wget -i download3.sh
for sample in $(cat /data/mice/PRJNA796303/unique_samples); do
fastq-dump --split-files --gzip $sample --outdir /data/mice/PRJNA796303/rawData
done
R1-barcode +UMI
R2-insert
I-sample index
cellranger was used to map reads to mouse genome:
for sample in $(cat /data/mice/PRJNA796303/unique_samples); do
cellranger count --id=$sample \
--fastqs=/data/mice/PRJNA796303/rawData \
--sample=$sample \
--no-bam \
--transcriptome=/data/mice/PRJNA796303/refdata-gex-GRCm39-2024-A;
done
SRR17555083
SRR17555084
SRR17555085
SRR17555086
SRR17555087
SRR17555088
SRR17555089
SRR17555090
cell ranger output files were transferred to local computer and analyzed in Seurat 5.1.0 (Bioconductor 3.19)
Analysis aligned data in Seurat
Loading required libraries
library(SCINA)
library(cerebroApp)
library(msigdb)
library(scmap)
library(celldex)
library(SingleR)
library(Seurat)
library(hdf5r)
library(gdata)
library(Matrix)
library(DropletUtils)
library(patchwork)
library(harmony)
library(SoupX)
library(glmnet)
library(ComplexHeatmap)
library(ggplot2)
library(dplyr)
library(EBImage)
library(glmGamPoi)
library(tibble)
library(repr)
library(purrr)
files <- list.files(path="/Users/alexeyefanov/Desktop/Programming/biobase/GSE193462/output",recursive=T,pattern="*.h5")
files
'D10/filtered_feature_bc_matrix.h5''D16/filtered_feature_bc_matrix.h5'
'D20/filtered_feature_bc_matrix.h5''D26/filtered_feature_bc_matrix.h5'
'WT10/filtered_feature_bc_matrix.h5''WT16/filtered_feature_bc_matrix.h5'
'WT20/filtered_feature_bc_matrix.h5''WT26/filtered_feature_bc_matrix.h5'
Reading hdf5 files (extension .h5)
The h5_read is the list of matrices.
h5_read <- lapply(paste0("/Users/alexeyefanov/Desktop/Programming/biobase/GSE193462/output/", files), Read10X_h5)
names(h5_read)<- sapply(files,
function(x){str_split_1(x,"/")[1]}
)
names(h5_read)
'D10''D16''D20''D26''WT10''WT16''WT20''WT26'Creating Seurat object
adp <- mapply(CreateSeuratObject,counts=h5_read,
project=names(h5_read),
MoreArgs = list(min.cells = 3, min.features = 200))
Merge Seurat objects
adp <- merge(adp[[1]], y = adp[2:length(adp)],
add.cell.ids = names(adp),project="Adipose")
adp
An object of class Seurat
24912 features across 49138 samples within 1 assay
Active assay: RNA (24912 features, 0 variable features)
8 layers present: counts.D10, counts.D16, counts.D20, counts.D26, counts.WT10, counts.WT16, counts.WT20, counts.WT26
slotNames(adp)
'assays''meta.data''active.assay''active.ident''graphs''neighbors''reductions''images''project.name''misc''version''commands''tools'
adp@assays
$RNA
Assay (v5) data with 24912 features for 49138 cells
First 10 features:
Xkr4, Gm1992, Gm19938, Rp1, Sox17, Mrpl15, A930006A01Rik, Lypla1,
Tcea1, Rgs20
Layers:
counts.D10, counts.D16, counts.D20, counts.D26, counts.WT10,
counts.WT16, counts.WT20, counts.WT26
head(adp@meta.data)
A data.frame: 6 × 3
orig.ident nCount_RNA nFeature_RNA
D10_AAACCCAAGATGCTTC-1 D10 13602 4182
D10_AAACCCAAGCTCTTCC-1 D10 38649 6789
D10_AAACCCAAGTCATCGT-1 D10 31695 5694
D10_AAACCCAGTAGCGTCC-1 D10 21310 4959
D10_AAACCCAGTGCACGCT-1 D10 28390 4977
D10_AAACCCAGTGTAAATG-1 D10 5835 2276
nCount_RNA - number of UMIs per cell
nFeature_RNA - number of genes expressed (detected) per cell
the meta.date data frame should be updated with type of mice (condition) and time points (time_point) information. The combination of condition and time_point will be added as well.
adp$condition <- ifelse(str_detect(adp@meta.data$orig.ident, "^W"),
"WT","DKO")
adp$time_point <- ifelse(str_detect(adp@meta.data$orig.ident, "0"),
"Day 0","Day 6")
adp$cond_tp <- paste(adp$condition, adp$time_point)Visualization of the number of cells per sample
adp@meta.data %>%
ggplot(aes(x=orig.ident, fill=orig.ident)) +
geom_bar(color="black") +
stat_count(geom = "text", colour = "black", size = 3.5,
aes(label = after_stat(count)),
position=position_stack(vjust=0.5))+
theme_classic() +
theme(plot.title = element_text(hjust=0.5, face="bold")) +
ggtitle("Number of Cells per Sample")
ggsave("seurat_barplot.png")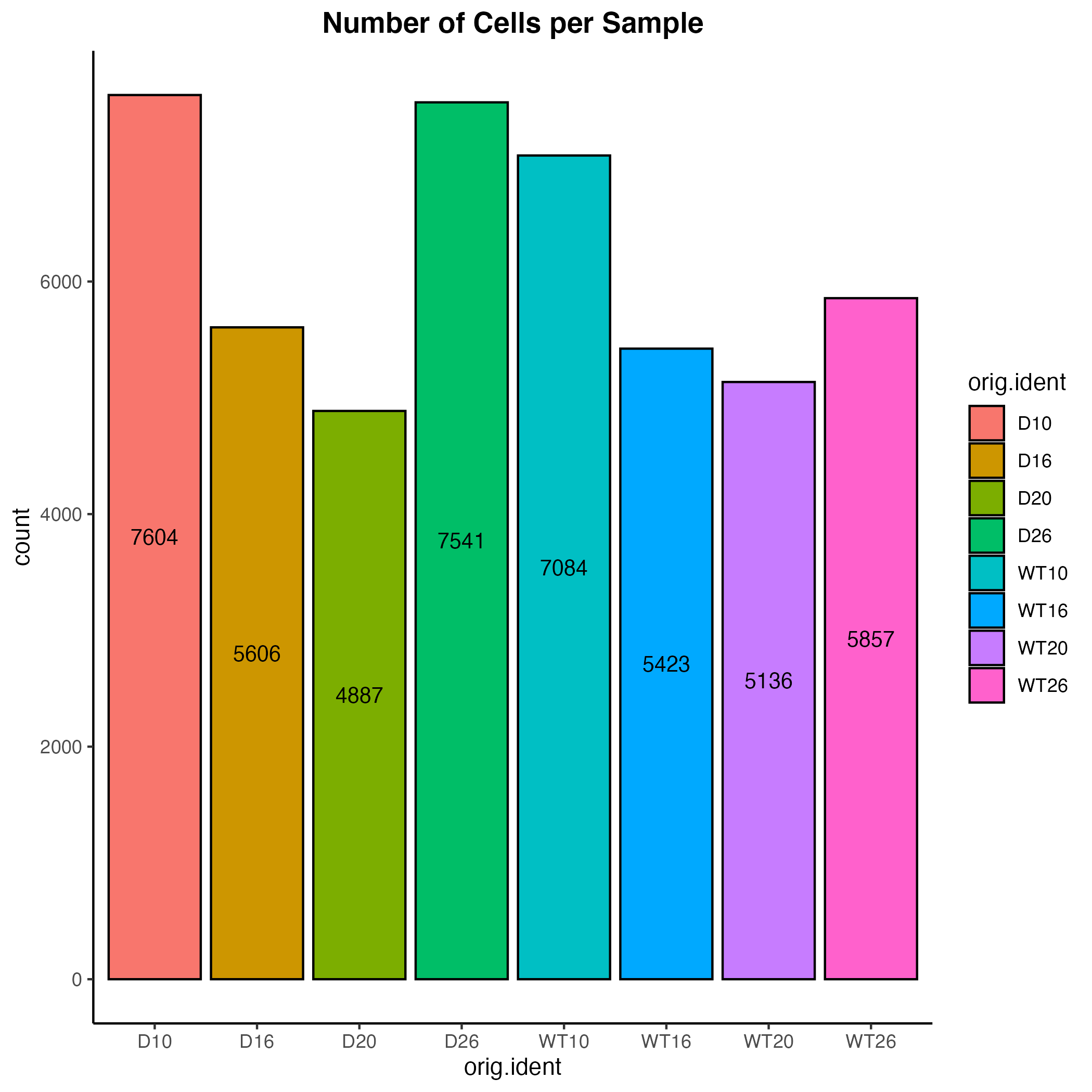
Saving adp object in R Data Serialization (RDS) format
saveRDS(adp,"/Users/alexeyefanov/Desktop/Programming/biobase/GSE193462/output/merged_Seurat_adp1.rds")Quality Control
There are 3 main QC metrics:- nCount_RNA
- nFeature_RNA
- percent.mt: the fraction of reads from mitochondrial genes per cell
adp[["percent.mt"]] <- PercentageFeatureSet(adp, pattern = "^mt-")
metadata <- adp@meta.data
cnames<-setNames(rep(c("cyan3","darkgoldenrod1"),each=4),levels(factor(metadata$orig.ident)))
levels(factor(metadata$orig.ident))
cnames
'D10''D16''D20''D26''WT10''WT16''WT20''WT26'
D10'cyan3'D16'cyan3'D20'cyan3'D26'cyan3'WT10'darkgoldenrod1'WT16'darkgoldenrod1'WT20'darkgoldenrod1'WT26'darkgoldenrod1'
VlnPlot(adp, features = "percent.mt", group.by="orig.ident") +
scale_fill_manual(values=cnames) +
geom_hline(yintercept=10,color="red")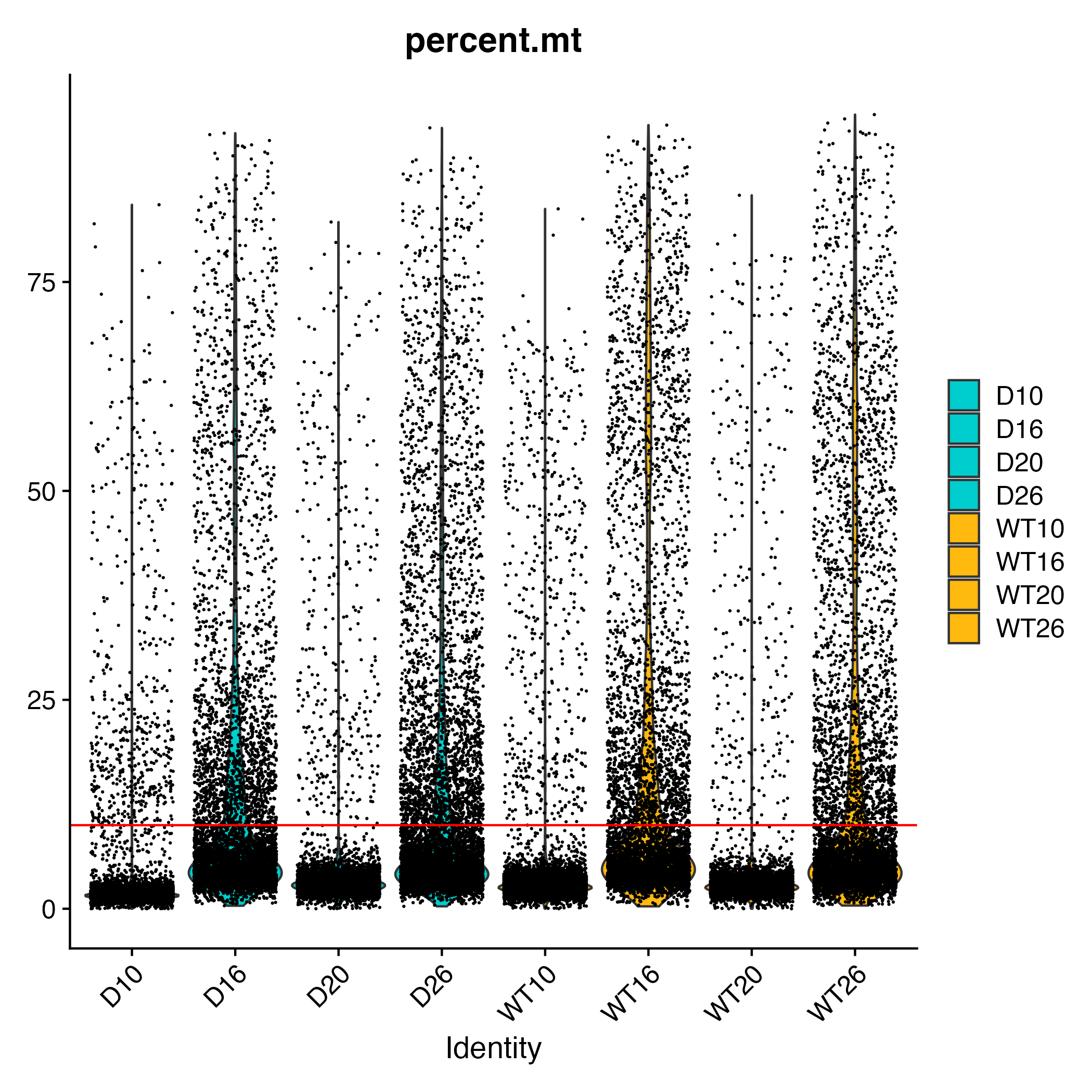
Selected filtration criteria:
Day 0:
nFeature_RNA > 350
nCount_RNA >650
percent.mt <10
Day 6:
nFeature_RNA > 350
nCount_RNA >650
percent.mt <25
t0<- metadata |> filter(time_point=="Day 0", nFeature_RNA > 350,
nCount_RNA >650, percent.mt <10 ) |>
rownames_to_column("Cell") |> pull(Cell)
t6<- metadata |> filter(time_point=="Day 6", nFeature_RNA > 350,
nCount_RNA >650, percent.mt <25 ) |>
rownames_to_column("Cell") |> pull(Cell)
keep<-c(t0,t6)Filtering the seurat object
adp_filt<-subset(adp, cells=keep)
saveRDS(adp_filt,"/Users/alexeyefanov/Desktop/Programming/biobase/GSE193462/output/adp_merge_filt1.rds")
Normalization
library(sctransform)
library(glmGamPoi)
adp_filt<-readRDS("/Users/alexeyefanov/Desktop/Programming/biobase/GSE193462/output/adp_merge_filt_sctran_clust0.1.rds")
Linear Dimensionality Reduction
adp_filt <- RunPCA(adp_filt, verbose = FALSE, assay="SCT")
Exploring the PCA results
options(repr.plot.width=10, repr.plot.height=10)
ElbowPlot(adp_filt, ndims = 40)
ggsave("elbow.jpg")
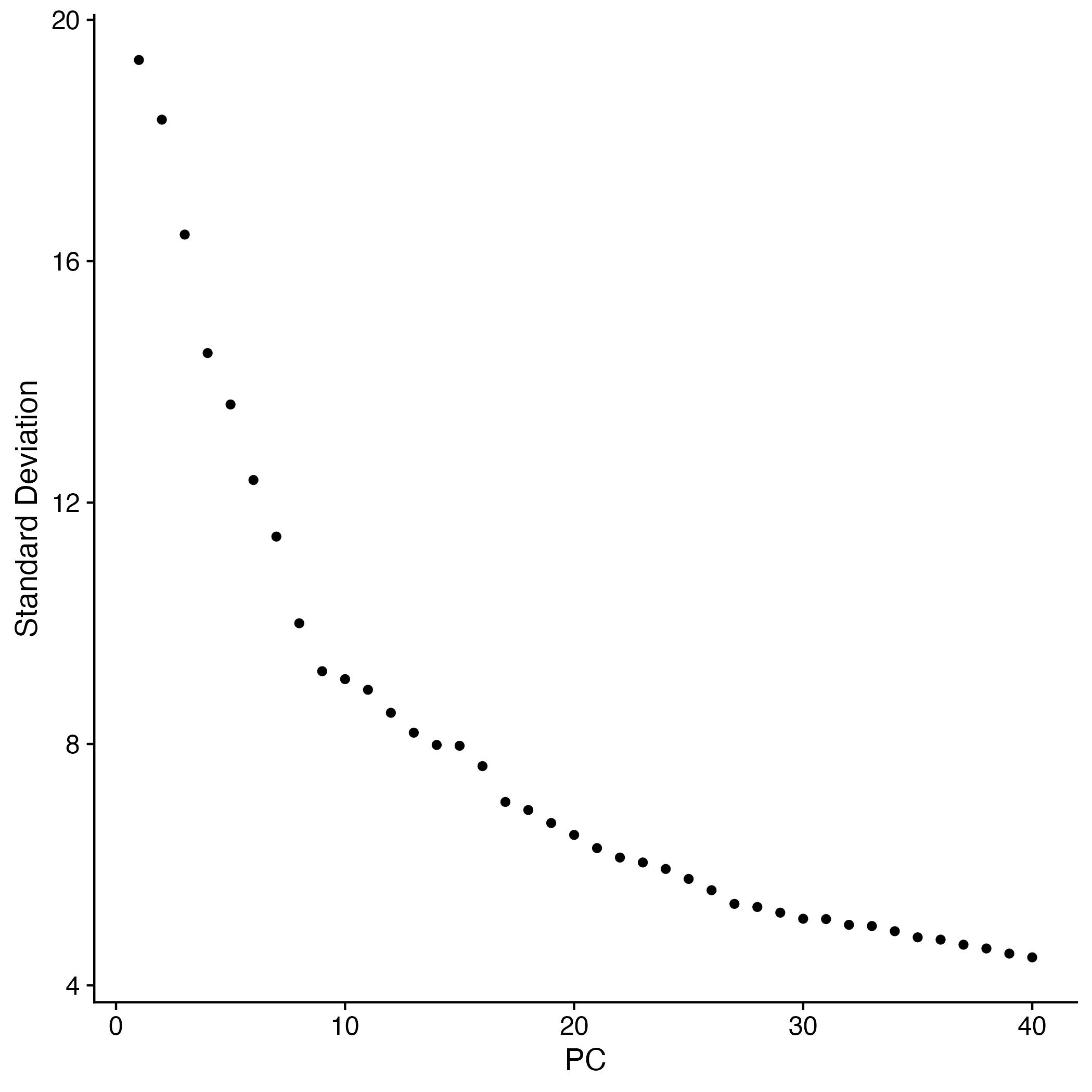
The majority of variation is captured by around 30 PC.
Clustering
adp_filt <- FindNeighbors(adp_filt, dims = 1:30)
adp_filt <- FindClusters(adp_filt, resolution = 0.1)
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 42502
Number of edges: 1423435
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9509
Number of communities: 9
Elapsed time: 8 seconds
Cluster visualization using uniform manifold approximation and projection (UMAP)
adp_filt <- RunUMAP(adp_filt,dims = 1:30)
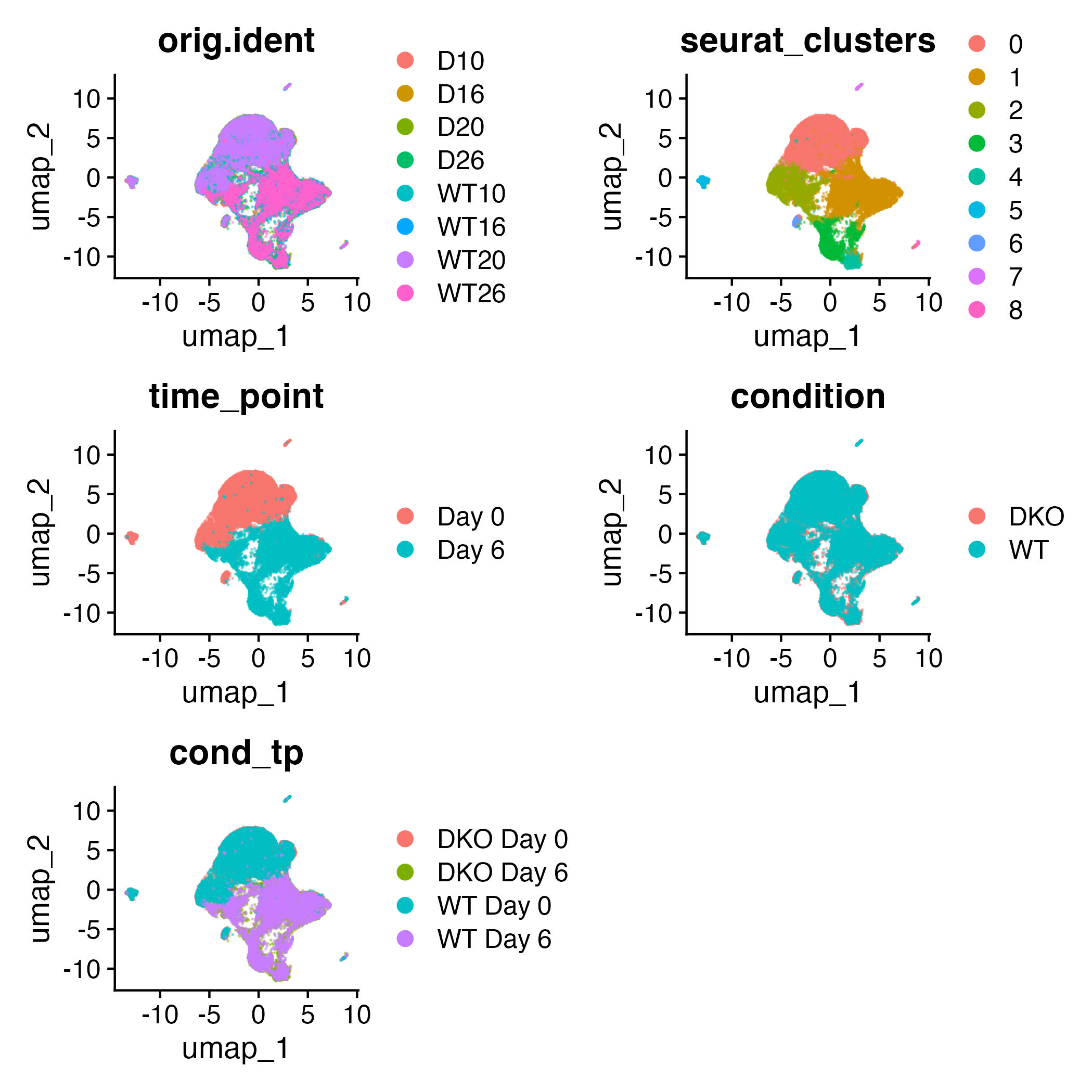
Visualizing potential contamination
Ptprc (CD45+): nucleated hematopoietic cellsPecam: endothelial cells
Plp1:neuronal and glial cells
contam=c("Ptprc", "Plp1", "Pecam1")
VlnPlot(adp_filt, features=contam)
ggsave("contam.jpg")
FeaturePlot(adp_filt,features=contam)
ggsave("contam_cluster.jpg")

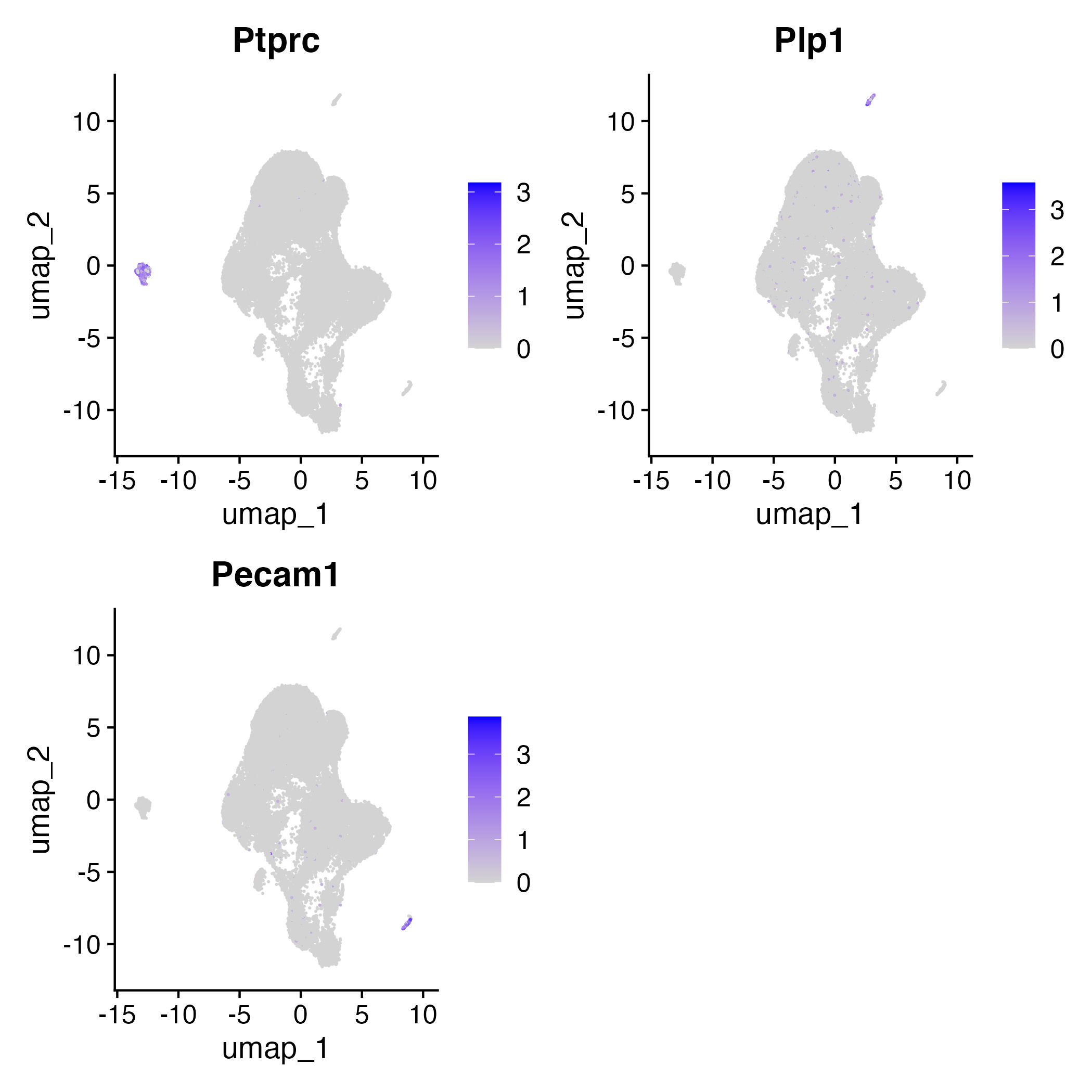
Removal clusters with high contaminations of Ptprc, Plp1, and Pecam1
adp<-adp_filt
Idents(adp) = "SCT_snn_res.0.1"
`%not_in%` <- purrr::negate(`%in%`)
adp <- subset(adp, subset=seurat_clusters %not_in% c(5,7, 8))
Finding Cluster Biomarkers
adp<-PrepSCTFindMarkers(adp, verbose=T)
Idents(adp) = "SCT_snn_res.0.1"
DefaultAssay(adp) = "SCT"
de_allClusters = FindAllMarkers(adp, test.use="wilcox", min.pct=0.1, logfc.threshold = 0.25, only.pos=TRUE)
Selecting 5 top differentially expressed (DE) genes in all clusters
de_allClusters %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
head(top5)
Heatmap of 5 top DE genes for each cluster
options(repr.plot.width=8, repr.plot.height=8)
DoHeatmap(adp,features = top5$gene, slot="scale.data")
ggsave("doheatmap.jpg")
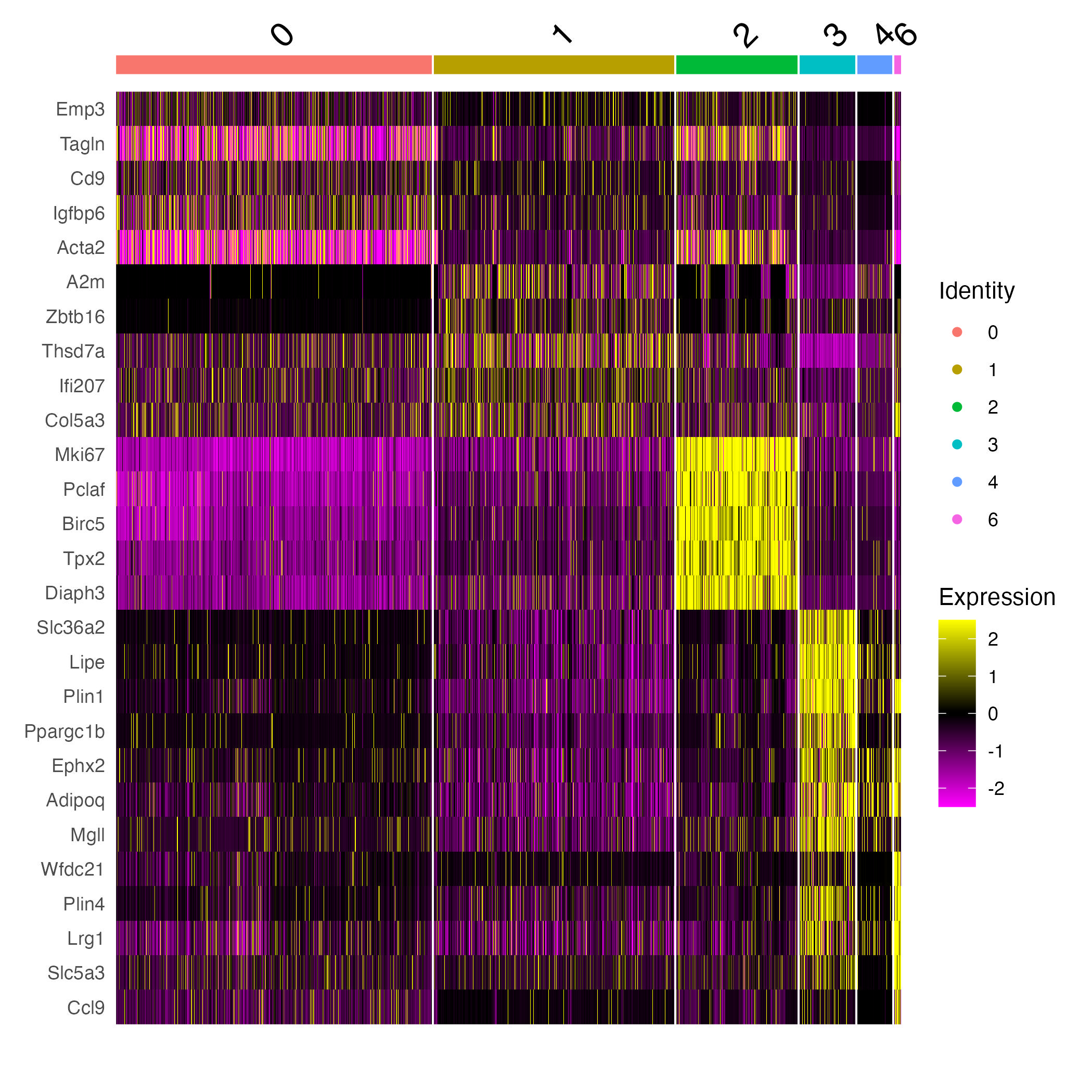
Finding markers between different time points (day 6 and day 0)
Idents(adp) = "time_point"
day6_day0_de=FindMarkers(adp,ident.1="Day 6",ident.2="Day 0",test.use="wilcox")
Visualizing clusters grouped by time points (day 6 and day 0)
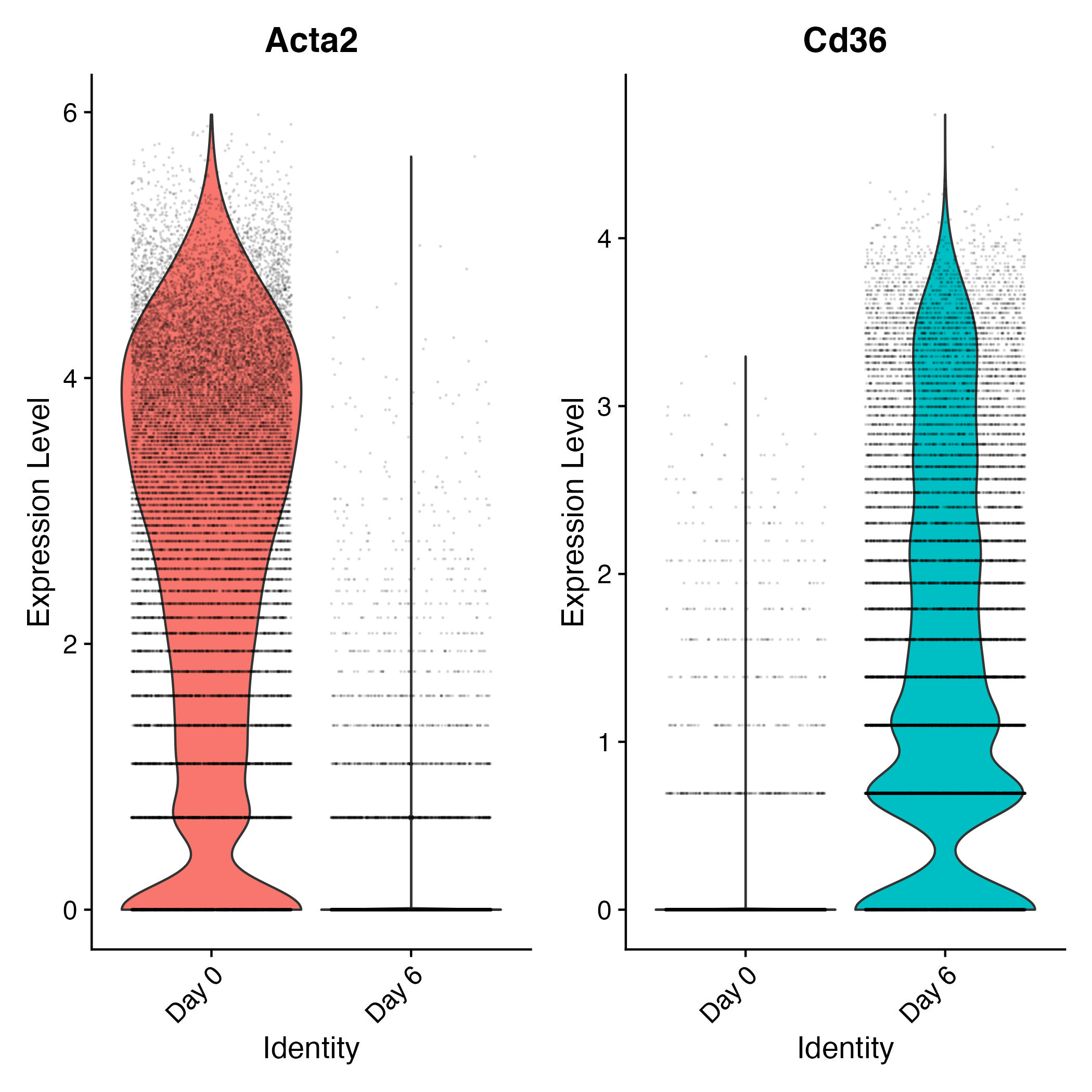
fig1 = DimPlot(adp,group.by="time_point")
fig2 = FeaturePlot(adp,features="Acta2",order=T)
fig3 = FeaturePlot(adp,features="Cd36",order=T)
fig1/(fig2|fig3)
ggsave("dimplot_time_point.jpg")
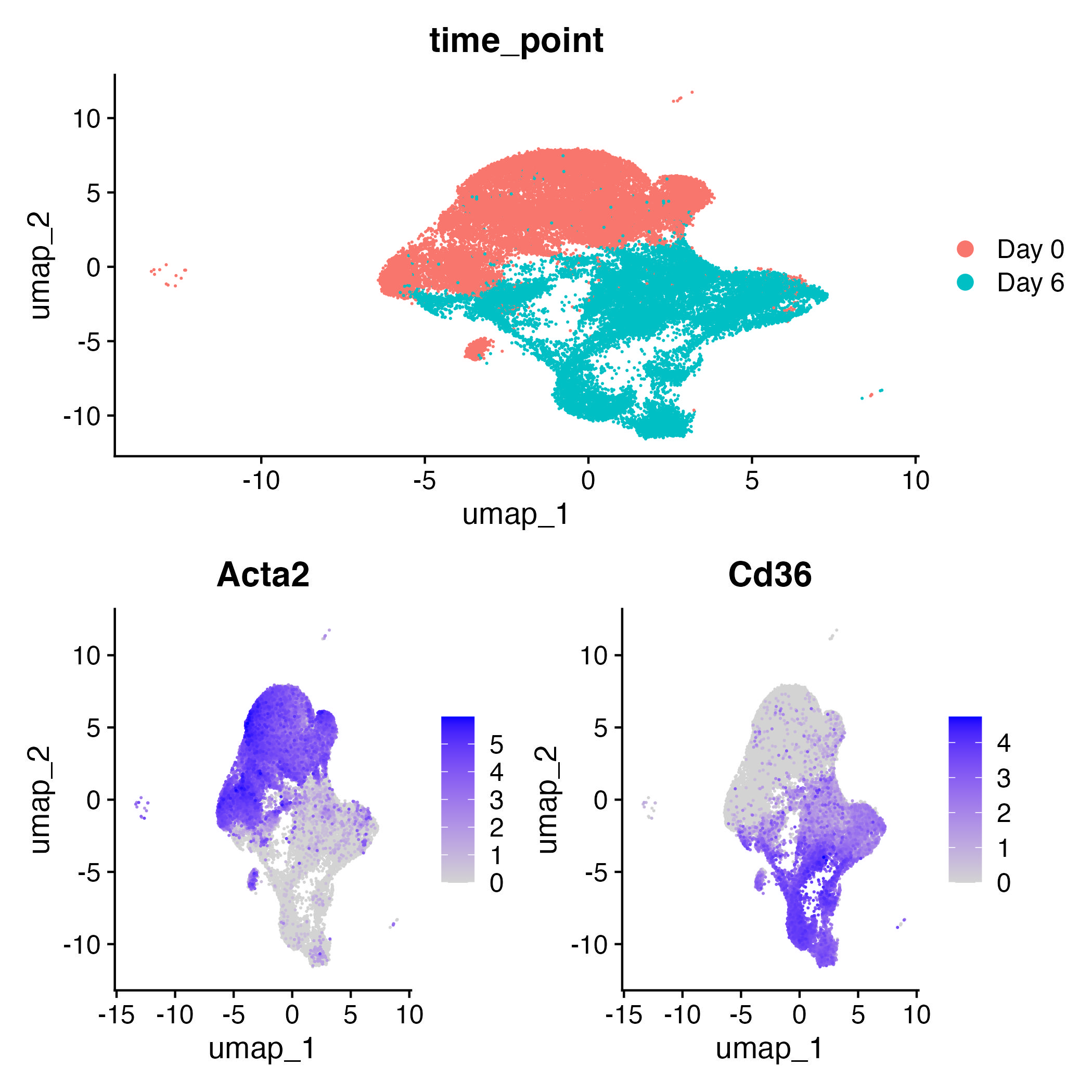
Acta2 is a marker of day 0, while cd36 is a marker of day 6.
Pseudobulk Differential Expression
The p-value inflation is a problematic point of scRNASeq analysis. The p-value inflation is a consequence of large sizes of compared samples. One approach to deal with the p-value inflation is to run a pseudobulk analysis.Pseudobulk Differential Expression
pseudo_adp=AggregateExpression(adp,assays="RNA", return.seurat=T, group.by=c("orig.ident","time_point","condition","cond_tp"))
Cleaning metadata of the pseudo_adp seurat object
pseudo_adp = RenameCells(pseudo_adp,new.names=gsub("_.*","",pseudo_adp$orig.ident))
pseudo_adp$orig.ident=gsub("_.*","",pseudo_adp$orig.ident)
Finding DE markers between different time points of Pseudobulk analysis
Idents(pseudo_adp)="time_point"
bulk_adp_de = FindMarkers(pseudo_adp, ident.1="Day 6", ident.2="Day 0", test.use="DESeq2")
Comparing the results between the single cell and pseudobulk analysis
scDE.genes = rownames(day6_day0_de)[which(day6_day0_de$p_val_adj<0.05)]
bulkDE.genes = rownames(bulk_adp_de)[which(bulk_adp_de$p_val_adj<0.05)]
length(scDE.genes)
length(bulkDE.genes)
length(intersect(scDE.genes,bulkDE.genes))
11306
10516
8503
Checking significance of DE expression in pseudobulk analysis for
bulk_adp_de[c("Acta2","Cd36"),]
p_val avg_log2FC pct.1 pct.2 p_val_adj
Acta2 1.256361e-274 -5.544797 1 1 3.129847e-270
Cd36 3.224132e-78 4.955947 1 1 8.031959e-74
Visuzlizing 5 top differentially expressed genes for each cluster in scRNAseq analysis
options(repr.plot.width=7, repr.plot.height=7)
Idents(adp)="SCT_snn_res.0.1"
DotPlot(adp,features=unique(top5$gene),dot.scale = 3)+coord_flip()
ggsave("dotplot_DE_genes.jpg")
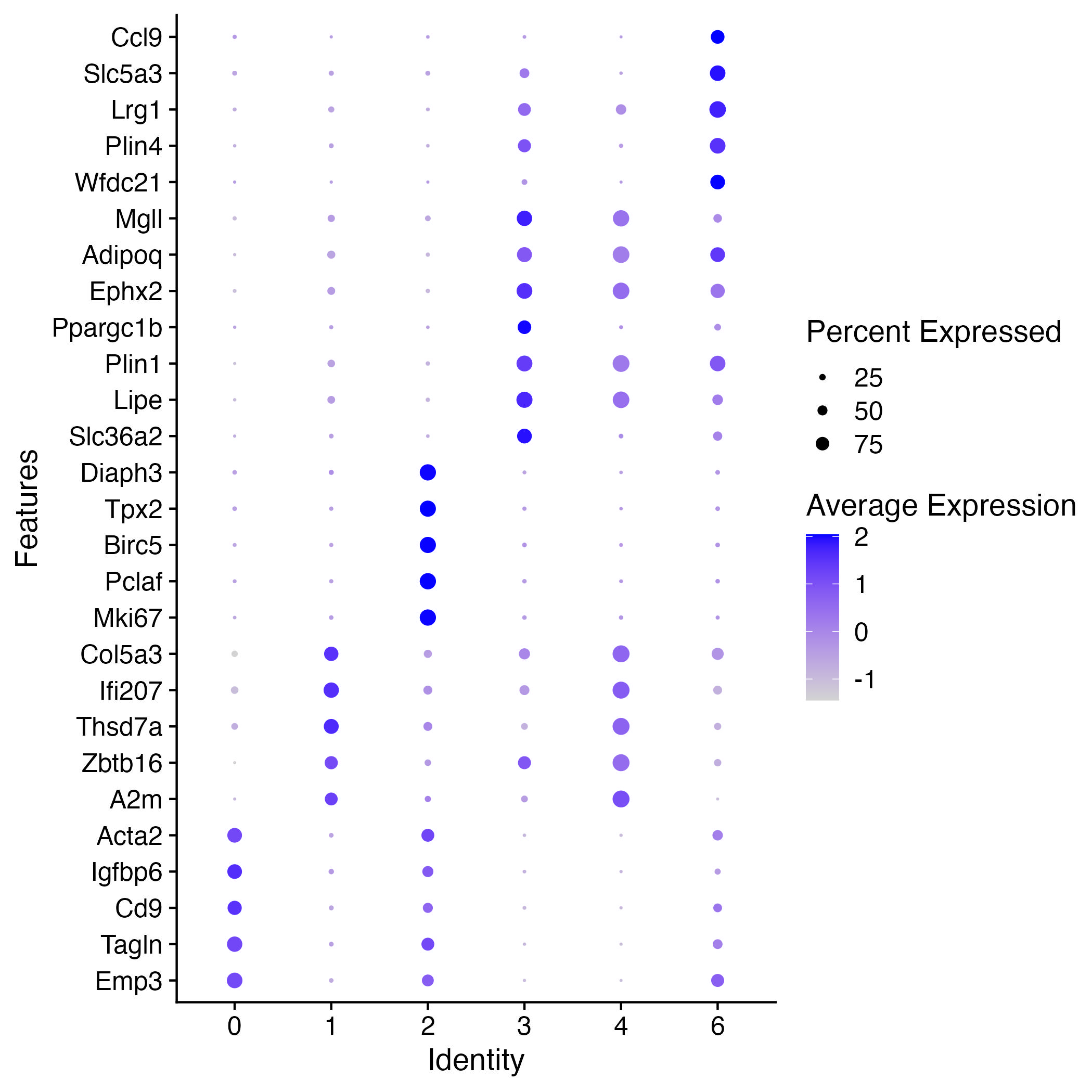
Another visualization of DE expressed genes is the violin plot
options(repr.plot.width=7, repr.plot.height=7)
Idents(adp) = "time_point"
VlnPlot(adp,features=c("Acta2","Cd36"),alpha = 0.1)
ggsave("violin_time_point.jpg")
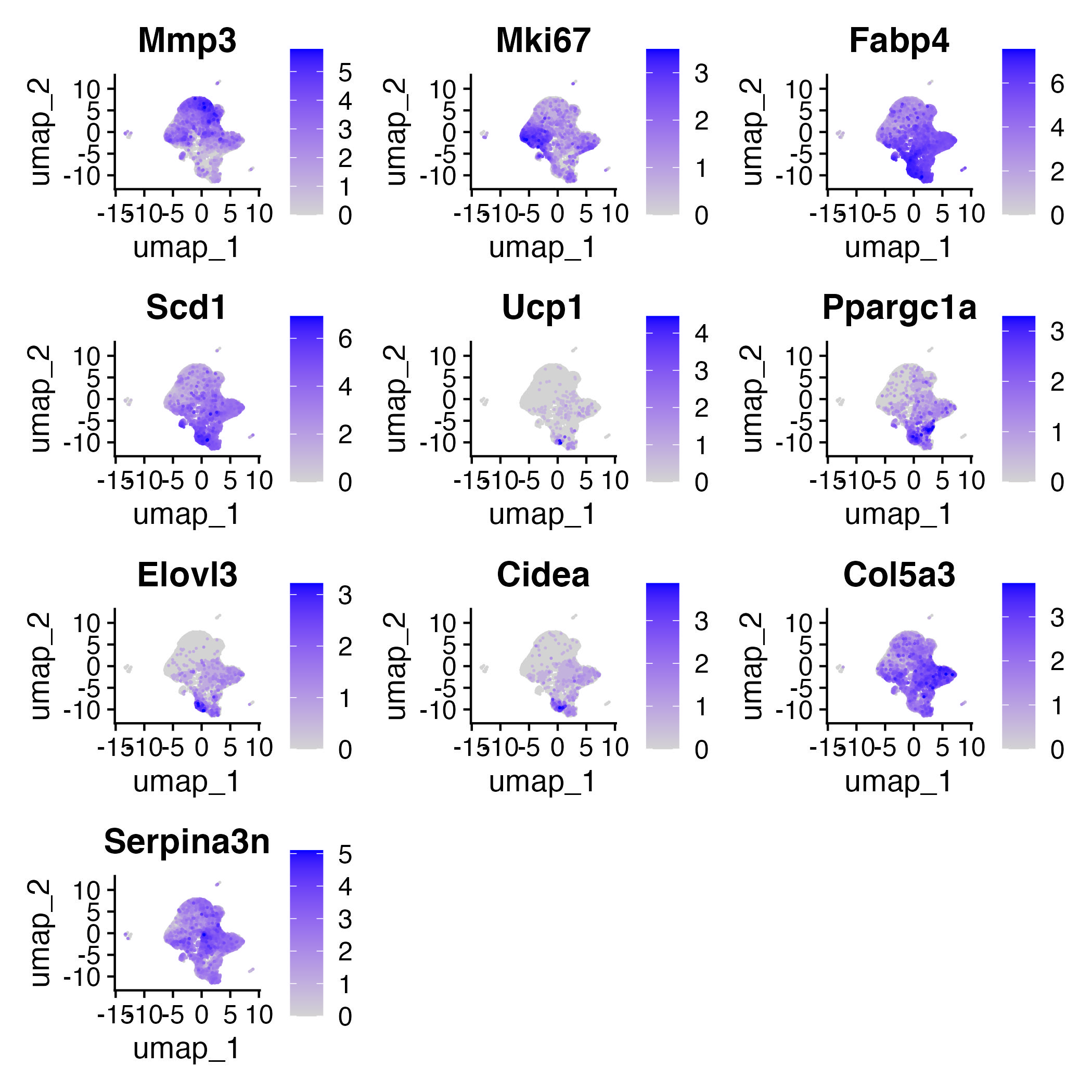
Cell anotation
Annotation by DE Markers was obtained from the paper:
Mmp3: preadipocytes
Mki67: proliferating cells
Fabp4: differentiating beige adipocytes and differentiated beige adipocytes
Scd1: differentiated beige adipocytes
Ucp1: differentiated beige adipocytes
Ppargc1a: differentiated beige adipocytes
Elovl3: differentiated beige adipocytes
Cidea: differentiated beige adipocytes
markers=c("Mmp3","Mki67","Fabp4","Scd1","Ucp1","Ppargc1a","Elovl3","Cidea", "Col5a3", "Serpina3n")
Idents(adp)="SCT_snn_res.0.1"
avgExp = AverageExpression(adp,markers,assay="SCT")$SCT
Cluster labeling
DimPlot(adp,label=T, label.size=6)
ggsave("dimplot_cluster_labels.jpg")

options(repr.plot.width=20, repr.plot.height=20)
FeaturePlot(adp,features=markers,ncol=3,order=T)
ggsave("Feature_plot_markers.jpg")
From the above graphs we can coclude:
Preadipocytes: Clusters 0 (Mp3)
Proliferating cells: Clusters 2, 6 (Mki67)
Differentiating beige adipocytes: Cluster 1
Differentiating adipocytes: Cluster 1, 4
options(repr.plot.width=10, repr.plot.height=12)
adipocyte=vector(length=ncol(adp))
adipocyte[which(adp$SCT_snn_res.0.1 %in% c(0))]="Preadipocytes"
adipocyte[which(adp$SCT_snn_res.0.1 %in% c(2,6))]="Proliferating cells"
adipocyte[which(adp$SCT_snn_res.0.1 %in% c(1,4))]="Differentiating adipocytes"
adipocyte[which(adp$SCT_snn_res.0.1 %in% c(3))]="Differentiated beige adipocytes"
adp$adipocyte = adipocyte
f1 = DimPlot(adp,group.by="SCT_snn_res.0.1",label=T) + NoLegend()
f2 = DimPlot(adp,group.by="time_point") + NoLegend()
f3 = DimPlot(adp,group.by="adipocyte",label=T)+NoLegend()
(f1|f2)/f3
ggsave("final_labelling.jpg")
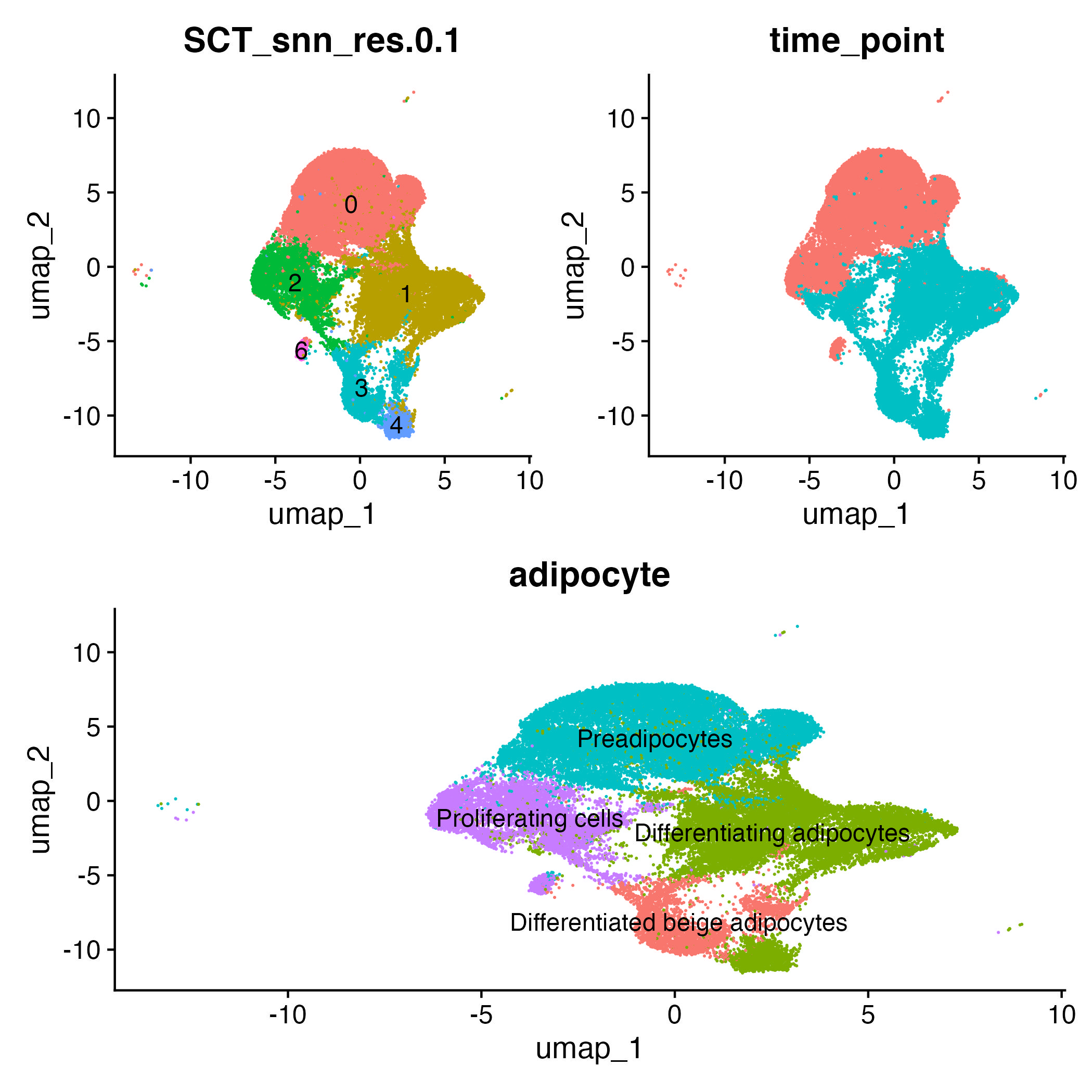
Conclusion: Preadipocytes and proliferating cells are mapped to day 0, while differentiated adipocytes, and differentiated beige adipocytes are mapped to day 6. There is a slight difference between the paper labeling and the current analysis labeling that can be attributed to the different reference transcriptome (2024 release year for the current analysis and 2020 release year in the paper analysis), cellranger and seurat versions.